ARASENS 試験:mCSPC 患者さんに対し、ADT、Darolutamide(ダロルタミド)、Docetaxel(ドセタキセル)を併用したTriplet レジメン
試験デザイン
無作為化・二重盲検・プラセボ対照・第Ⅲ相試験
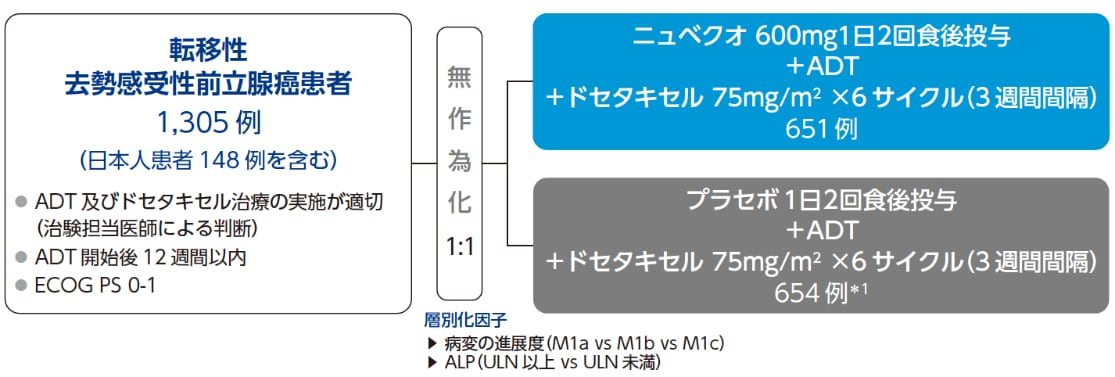
目的 |
標準的アンドロゲン遮断療法(ADT)とドセタキセルの併用にニュベクオ又はプラセボを上乗せした際のニュベクオの全生存期間(OS)の優越性を検討する。 |
|---|---|
対象 |
ADT開始後12週間以内の転移性*2去勢感受性前立腺癌患者 |
投与方法 |
ニュベクオ群又はプラセボ群に1:1の割合で無作為化し、ADTとドセタキセル(6サイクル)併用下で盲検にて投与した。
|
評価項目 |
|
解析計画 |
|
- *1: プラセボ群に無作為割付けされた654例のうち3例は、投与を受けなかった。
- *2: 骨シンチグラフィー陽性、若しくは、軟部組織転移又は内臓転移については腹部/骨盤/胸部造影CT又はMRI検査により放射線学的に転移が確定された患者を適格とした。所属リンパ節以外のリンパ節転移(M1a)、及び/又は骨転移(M1b)、及び/又は骨転移の有無を問わず他の部位に遠隔転移(M1c)を認める患者のみを適格とした[所属リンパ節転移のみ(N1、大動脈分岐部の下部)の患者は除外]。
- *3: 第一世代抗アンドロゲン剤の併用/非併用を問わずADT(LHRHアゴニスト/アンタゴニスト又は精巣摘除術)を無作為割付け前12週間以内に開始していること。LHRHアゴニスト投与患者では、第一世代抗アンドロゲン剤を無作為割付け前4週間以上併用することが推奨された。ただし、第一世代抗アンドロゲン剤は無作為割付け時には中止していること。
- *4: ドセタキセルは試験薬投与開始後6週間以内に開始し、治験担当医師の判断でprednisone(国内未承認)/プレドニゾロンと併用投与可とした。
SSE-FS: Symptomatic Skeletal Event-Free Survival
ULN: Upper Limit of Normal
- 6) バイエル薬品社内資料 [転移性去勢感受性前立腺癌患者を対象とした国際共同第Ⅲ相臨床試験(試験17777)] 承認時評価資料
- 7) バイエル薬品社内資料 [試験17777: 有効性評価項目]承認時評価資料
- 8) バイエル薬品社内資料 [試験17777: 安全性評価項目]承認時評価資料
- 9) Smith MR, et al.: N Engl J Med., 386; 1132-1142(2022) 本研究はバイエル、共同開発したOrion Corporation Orion Pharma(Orion社)の資金により実施された。著者には、バイエル、Orion社から講演料・コンサルタント料等を受領した者、およびバイエルの社員3名、Orion社の社員1名が含まれる。
患者背景
解析対象となった1,305例の患者背景は、以下のとおりです。
なお、アジア人の475例のうち、日本人は148例でした。
| ニュベクオ群 (ニュベクオ+ADT+ドセタキセル) (n=651) | プラセボ群 (プラセボ+ADT+ドセタキセル) (n=654) | ||
|---|---|---|---|
| 年齢 中央値[範囲] | 67.0歳[41~89] | 67.0歳[42~86] | |
| 年齢群 | < 65歳 65~74歳 75~84歳 ≧ 85歳 | 243(37.3%) 303(46.5%) 102(15.7%) 3(0.5%) | 234(35.8%) 306(46.8%) 110(16.8%) 4(0.6%) |
| 地域 | 北米 アジア太平洋 その他 | 125(19.2%) 229(35.2%) 297(45.6%) | 119(18.2%) 244(37.3%) 291(44.5%) |
| 人種 | 白人 黒人/アフリカ系 アジア人 その他/報告なし | 345(53.0%) 26(4.0%) 230(35.3%) 50(7.7%) | 333(50.9%) 28(4.3%) 245(37.5%) 48(7.3%) |
| ECOG PS | 0 1 不明 | 466(71.6%) 185(28.4%) 0 | 462(70.6%) 190(29.1%) 2(0.3%) |
| 初回診断時のGleasonスコア | < 8 ≧ 8 不明 | 122(18.7%) 505(77.6%) 24(3.7%) | 118(18.0%) 516(78.9%) 20(3.1%) |
| 初回診断時の転移 | あり なし 不明 | 558(85.7%) 86(13.2%) 7(1.1%) | 566(86.5%) 82(12.5%) 6(0.9%) |
| ベースライン時の 病変の進展度 | M1a(非所属リンパ節転移のみ) M1b(骨転移±リンパ節転移) M1c(内臓転移±リンパ節転移又は骨転移) | 23 (3.5%) 517(79.4%) 111(17.1%) | 16 (2.4%) 520(79.5%) 118(18.0%) |
| ベースライン時のPSA値 中央値[範囲] | 30.30ng/mL[0.0~9219.0] | 24.20ng/mL[0.0~11947.0] | |
| ベースライン時のALP値*1中央値[範囲] | 148.5U/L[40~4885]*2 | 140.0U/L[36~7680]*3 | |
| ベースライン時のALP値 | <ULN ≧ULN | 290(44.5%) 361(55.5%) | 291(44.5%) 363(55.5%) |
*1: IFCC法で測定した。 *2: n=652 *3: n=650
全生存期間(OS)【主要評価項目、検証的解析結果】
ニュベクオ群のプラセボ群に対するハザード比は0.675〔95%CI: 0.568~0.801〕、p<0.0001であり、OSについて、優越性が検証されました。OSの中央値は、ニュベクオ群で未到達、プラセボ群で48.9ヵ月でした。48 ヵ月時点での生存率は、ニュベクオ群で62.7%、プラセボ群で50.4%でした。

層別化因子:病変の進展度(M1a vs M1b vs M1c)、ALP(ULN 以上 vs ULN 未満)
OS:無作為割付け日からあらゆる原因による死亡までの期間と定義した。死亡が確認されていない被験者のOS は、生存が確認された最後の日又はデータベースのカットオフ日のいずれか早い方で打ち切った。
追跡期間中央値:ニュべクオ群43.7ヵ月、プラセボ群42.4ヵ月(データカットオフ:2021年10月25日)
OSのサブグループ解析
事前に規定された全てのサブグループにおいて、点推定値が1を下回りました。

層別化をしていない単変量でのCox回帰モデルによる。
ULN: Upper Limit of Normal
CRPCとなるまでの期間【副次評価項目】
CRPCとなるまでの期間の中央値は、ニュベクオ群で未到達、プラセボ群で19.1ヵ月でした。ニュベクオ 群のプラセボ群に対するハザード比は0.357〔95%CI: 0.302~0.421〕、p<0.0001であり、ニュベクオ群でCRPCとなるまでの期間の有意な延長が認められました。

層別化因子:病変の進展度(M1a vs M1b vs M1c)、ALP(ULN 以上 vs ULN 未満)
CRPCとなるまでの期間:無作為割付け日からのPSA無増悪期間(血清中テストステロンが去勢レベルの0.50ng/mL未満の状態でのPSA増悪が最初に認められるまでの期間)、又は軟部組織/内臓の病変に基づく無増悪期間、又は骨病変に基づく無増悪期間のうち、いずれか短い方の期間と定義した。
- PSA増悪とは、PCWG3基準に従い、ベースライン時又はベースライン後の最低値から変化率で25%以上、かつ実測値で2ng/mL以上の上昇を認めた場合と定義した。
- 軟部組織/内臓の病変に基づく無増悪期間は、治験担当医師が行う胸部、腹部及び骨盤のCT/MRI検査に基づき、PCWG3基準に従い、RECIST ver1.1に準じて判定した。
- 骨病変に基づく無増悪期間は、治験担当医師が行う99mTcメチレンジホスホン酸を用いた全身骨シンチグラフィーに基づき、PCWG3基準に従い判定した。
PCWG 3:Prostate Cancer Clinical Trials Working Group 3
追跡期間中央値:ニュべクオ群43.7ヵ月、プラセボ群42.4ヵ月(データカットオフ:2021年10月25日)
後治療開始までの期間【副次評価項目】
後治療開始までの期間の中央値は、ニュベクオ群で未到達、プラセボ群で25.3ヵ月でした。ニュベクオ群のプラセボ群に対するハザード比は0.388〔95%CI: 0.328~0.458〕、p<0.0001であり、ニュベクオ群で後治療開始までの期間の有意な延長が認められました。

層別化因子:病変の進展度(M1a vs M1b vs M1c)、ALP(ULN 以上 vs ULN 未満)
後治療開始までの期間:無作為割付けから、前立腺癌に対する後治療の開始までの期間と定義した。
追跡期間中央値:ニュべクオ群43.7ヵ月、プラセボ群42.4ヵ月(データカットオフ:2021年10月25日)
延命治療のための全身性抗腫瘍療法による後治療
- 延命治療のための全身性抗腫瘍療法による後治療は以下のとおりでした。
| n (%) | ニュベクオ群 (ニュベクオ+ADT+ドセタキセル) | プラセボ群 (プラセボ+ADT+ドセタキセル) | |
|---|---|---|---|
| アクティブフォローアップ期間または長期フォローアップ期間移行例*1 | 315/651(48.4) | 495/654(75.7) | |
| 延命のための後治療を受けた患者*2 1種類 2種類以上 | 179/315(56.8) 108/179(60.3) 71/179(39.7) | 374/495(75.6) 221/374(59.1) 153/374(40.9) | |
| アビラテロン | 112/315(35.6) | 232/495(46.9) | |
| エンザルタミド | 48/315(15.2) | 136/495(27.5) | |
| カバジタキセル | 57/315(18.1) | 89/495(18.0) | |
| ドセタキセル | 46/315(14.6) | 89/495(18.0) | |
| 塩化ラジウム[223Ra] | 19/315(6.0) | 34/495(6.9) | |
| Sipuleucel-T(国内未承認) | 4/315(1.3) | 10/495(2.0) | |
| Lutetium[177Lu] (国内未承認) | 1/315(0.3) | 6/495(1.2) | |
| アパルタミド | 2/315(0.6) | 2/495(0.4) | |
| [177Lu] ルテチウム-PSMA-617 | 1/315(0.3) | 1/495(0.2) | |
- *1: ニュベクオ群の患者1例はフォローアップ期間に移行しなかったが、後治療を受けた。
- *2: アビラテロン、アパルタミド、エンザルタミド、ドセタキセル、カバジタキセル、塩化ラジウム、Sipuleucel-T、およびルテチウムが延命治療として定義された。
- ※ データカットオフ時点で、ニュベクオ群299 例(45.9%)、プラセボ群125 例(19.1%)が試験薬の投与を継続中であった。試験薬の投与を中止した被験者は、ニュベクオ群352 例(54.1%)、プラセボ群526 例(80.4%)であった。
副次評価項目とその他の評価項目
ニュベクオ群、プラセボ群における、副次評価項目とその他の評価項目の結果は、次のとおりでした。
| ニュベクオ群(ニュベクオ+ADT+ドセタキセル) (n=651) |
プラセボ群(プラセボ+ADT+ドセタキセル) (n=654) |
ハザード比 〔95%CI〕 |
p値*1 | ||||
|---|---|---|---|---|---|---|---|
| 中央値 〔95%CI〕 |
イベント数 | 中央値 〔95%CI〕 |
イベント数 | ||||
| 副次評価項目 | CRPCとなるまでの期間 | 未到達 〔未到達~未到達〕 |
225 | 19.1ヵ月 〔16.5~21.8〕 |
391 | 0.357 〔0.302~0.421〕 |
<0.0001 |
| 疼痛増悪までの期間 | 未到達 〔30.5~未到達〕 |
222 | 27.5ヵ月 〔22.0~36.1〕 |
248 | 0.792 〔0.660~0.950〕 |
0.0058 | |
| SSE-FS | 51.2ヵ月 〔47.2~未到達〕 |
257 | 39.7ヵ月 〔36.0~42.3〕 |
329 | 0.609 〔0.516~0.718〕 |
<0.0001 | |
| SSEの初回発現までの期間 | 未到達 〔未到達~未到達〕 |
95 | 未到達 〔未到達~未到達〕 |
108 | 0.712 〔0.539~0.940〕 |
0.0081 | |
| 後治療開始までの期間 | 未到達 〔未到達~未到達〕 |
219 | 25.3ヵ月 〔23.1~28.8〕 |
395 | 0.388 〔0.328~0.458〕 |
<0.0001 | |
| 疾患の⾝体症状の 悪化までの期間 |
19.3ヵ月 〔13.8~24.8〕 |
351 | 19.4ヵ月 〔15.4~27.6〕 |
308 | 1.043 〔0.894~1.217〕 |
0.7073 | |
| 7⽇間以上連続する オピオイド使用の開 始までの期間*2 |
未到達 〔未到達~未到達〕 |
92 | 未到達 〔未到達~未到達〕 |
117 | 0.688 | ー | |
| その他の評価項目*3 | PSA増悪までの期間 | 未到達 〔未到達~未到達〕 |
136 | 22.4ヵ月 〔22.1~27.6〕 |
310 | 0.255 | ー |
- *1: 層別log-rank test(副次評価項目の有意水準:片側0.025)
- *2: 副次評価項目の解析には、逐次的ゲートキーピング法が用いられた。今回の解析では、疾患の身体症状の悪化までの期間が事前に規定した有意水準(片側0.025)を満たさなかったため、7日間以上連続するオピオイド使用の開始までの期間の検定は行わず、ハザード比のみを記載した。
- *3: その他の評価項目の検定結果は参考値であるため、ハザード比のみを記載した。
層別化因子:病変の進展度(M1a vs M1b vs M1c)、ALP(ULN 以上 vs ULN 未満)
疾患の身体症状の悪化までの期間:無作為割付けから、NCCN-FACT FPSI-17質問票に基づく身体的症状の悪化を最初に経験するまでの期間と定義した。疾患の身体症状の悪化は、4週間以上あけた2回の連続する評価で、疾患の身体症状サブスケール(FPSI-DRS-Pサブスケール)がベースラインから3ポイント低下すること(スコアが低いほど症状負荷が高いことを示す)と定義した。
7日間以上連続するオピオイド使用の開始までの期間:無作為割付けから、7日以上連続するオピオイド使用の開始までの期間と定義した。
NCCN-FACT FPSI-17 :Functional assessment of cancer therapy / National Comprehensive Cancer Network prostate cancer symptom index 17 item questionnaire
追跡期間中央値:ニュべクオ群43.7ヵ月、プラセボ群42.4ヵ月(データカットオフ:2021年10月25日)
PSA奏効率(無作為割付け後12ヵ月時点)【その他の評価項目】
- 相対的PSA奏効率は、ニュベクオ群で84.3%、プラセボ群で57.5%であった
- 絶対的PSA奏効率は、ニュベクオ群で60.2%、プラセボ群で26.1%であった
相対的PSA奏効率
(ベースラインから90%以上のPSA値の低下)
【その他の評価項目】")
絶対的PSA奏効率
(PSA値が0.2ng/mL未満)
【その他の評価項目】")
- 相対的PSA奏効率: 相対的PSA奏効(ベースラインから30%以上、50%以上及び90%以上のPSA値の低下)は、初回から3週間以降に行う2回目のPSA測定で確認した。
相対的PSA奏効率を、無作為割付け後3、6及び12ヵ月後までの被験者データに基づき評価した。 - 絶対的PSA奏効率: 絶対的PSA奏効(PSA値が0.2ng/mL未満)は、初回から3週後以降に行う2回目のPSA測定で確認した。
絶対的PSA奏効率を、無作為割付け後6及び12ヵ月後までの被験者データに基づき評価した。
有害事象の概要 [因果関係を問わない事象]
- 安全性解析対象集団における有害事象は、下記のとおりでした。
【試験薬投与期間中央値】ニュベクオ群:41.0ヵ月、プラセボ群:16.7ヵ月
n (%)
グレードはNCI-CTCAE Ver 4.03に準じる。 | ニュベクオ群 (ニュベクオ+ADT+ドセタキセル) (n=652*1) | プラセボ群 (プラセボ+ADT+ドセタキセル) (n=650*2) | |
|---|---|---|---|
| 全グレードの有害事象 | 649(99.5) | 643(98.9) | |
| グレード 3/4の有害事象 | 433(66.4) | 413(63.5) | |
| グレード 5の有害事象 | 27(4.1) | 26(4.0) | |
| 重篤な有害事象 | 293(44.9) | 275(42.3) | |
| 試験薬の投与中止に至った有害事象 | 89(13.7) | 69(10.6) | |
| ドセタキセルの投与中止に至った有害事象 | 52(8.0) | 67(10.3) | |
- ※ 上記表の安全性データを当局へ提出後、当局からの照会事項に基づき、プラセボ群の2例 [悪心、背部痛 各1例](いずれも因果関係は否定された)を追加報告している
- *1: プラセボ群に無作為化されたがニュベクオを投与された1例はニュベクオ群の安全性解析対象集団とされた。
- *2: プラセボ群に無作為化されたが治療を受けなかった3例は、安全性解析対象集団から除外された。
・ 主な重篤な有害事象(1%以上)
[ニュベクオ群: 発熱性好中球減少症 40例(6.1%)、好中球数減少 18例(2.8%)、肺炎 16例(2.5%)、好中球減少症 12例(1.8%)、発熱 9例(1.4%)、尿路感染 7例(1.1%)、COVID-19肺炎 7例(1.1%)]
[プラセボ群: 発熱性好中球減少症 39例(6.0%)、肺炎 21例(3.2%)、発熱 15例(2.3%)、好中球減少症 14例(2.2%)、好中球数減少 10例(1.5%)、アラニンアミノトランスフェラーゼ増加 8例(1.2%)、尿路感染 7例(1.1%)、脊髄圧迫 7例(1.1%)]
・ 主なニュベクオ又はプラセボの投与中止に至った有害事象(3例以上)
[ニュベクオ群: アスパラギン酸アミノトランスフェラーゼ増加 6例(0.9%)、アラニンアミノトランスフェラーゼ増加 5例(0.8%)、COVID-19肺炎 4例(0.6%)、斑状丘疹状皮疹 3例(0.5%)]
[プラセボ群: 骨痛 9例(1.4%)、肺炎 3例(0.5%)、背部痛 3例(0.5%)、間質性肺疾患 3例(0.5%)]
・主な死亡に至った有害事象(2例以上)
[ニュベクオ群: COVID-19肺炎 4例(0.6%)、突然死 2例(0.3%)]
[プラセボ群: 全身健康状態悪化 4例(0.6%)、突然死 3例(0.5%)、心停止 2例(0.3%)、死亡 2例(0.3%)、肺炎 2例(0.3%)、肺敗血症 2例(0.3%)]
主な有害事象 [因果関係を問わない事象](いずれかの群で15%以上発現)
データカットオフ:2021年10月25日
- いずれかの群で25%以上の発現割合でみられた有害事象は、脱毛症、疲労、貧血、 関節痛、末梢性浮腫、好中球数減少および下痢でした。
- ニュベクオ群でプラセボ群に比べて発現割合が3%以上高い有害事象は、貧血、食欲減退、高血圧*1、四肢痛、AST増加*2でした。
【試験薬投与期間中央値】 ニュベクオ群:41.0ヵ月、プラセボ群:16.7ヵ月
| MedDRA Ver.25.0 n(%) | ニュベクオ群(ニュベクオ+ADT+ドセタキセル)(n=652) | プラセボ群(プラセボ+ADT+ドセタキセル)(n=650) | ||||
|---|---|---|---|---|---|---|
| 全グレード | グレード3以上 | EAIR全グレード (100人年) | 全グレード | グレード3以上 | EAIR全グレード (100人年) | |
| 全有害事象 | 649(99.5) | 460(70.6) | ― | 643(98.9) | 439(67.5) | ― |
| 脱毛症 | 266(40.8) | 1 (0.2) | 15.4 | 264(40.6) | 2 (0.3) | 22.0 |
| 疲労 | 221(33.9) | 11 (1.7) | 12.8 | 216(33.2) | 12 (1.8) | 18.0 |
| 貧血 | 185(28.4) | 31 (4.8) | 10.7 | 164(25.2) | 33 (5.1) | 13.6 |
| 関節痛 | 182(27.9) | 8 (1.2) | 10.5 | 176(27.1) | 9 (1.4) | 14.6 |
| 末梢性浮腫 | 175(26.8) | 3 (0.5) | 10.1 | 170(26.2) | 1 (0.2) | 14.1 |
| 好中球数減少 | 171(26.2) | 152(23.3) | 9.9 | 155(23.8) | 140(21.5) | 12.9 |
| 下痢 | 169(25.9) | 8 (1.2) | 9.8 | 157(24.2) | 7 (1.1) | 13.1 |
| 白血球数減少 | 155(23.8) | 110(16.9) | 9.0 | 143(22.0) | 97(14.9) | 11.9 |
| 便秘 | 149(22.9) | 2 (0.3) | 8.6 | 131(20.2) | 2 (0.3) | 10.9 |
| 背部痛 | 127(19.5) | 12 (1.8) | 7.3 | 122(18.8) | 11 (1.7) | 10.1 |
| ほてり | 127(19.5) | 0 | 7.3 | 122(18.8) | 1 (0.2) | 10.1 |
| 食欲減退 | 121(18.6) | 1 (0.2) | 7.0 | 86(13.2) | 4 (0.6) | 7.2 |
| 悪心 | 117(17.9) | 3 (0.5) | 6.8 | 133(20.5) | 2 (0.3) | 11.1 |
| 体重増加 | 116(17.8) | 14 (2.1) | 6.7 | 105(16.2) | 8 (1.2) | 8.7 |
| ALT増加 | 103(15.8) | 18 (2.8) | 5.9 | 84(12.9) | 11 (1.7) | 7.0 |
| 四肢痛 | 102(15.6) | 2 (0.3) | 5.9 | 78(12.0) | 2 (0.3) | 6.5 |
- ※ 上記表の安全性データを当局へ提出後、当局からの照会事項に基づき、プラセボ群の2例 [悪心、背部痛 各1例](いずれも因果関係は否定された)を追加報告している
EAIR:曝露期間で調整した発現割合 ALT:アラニンアミノトランスフェラーゼ AST:アスパラギン酸アミノトランスフェラーゼ グレードはNCI-CTCAE Ver 4.03に準じる。
*1:ニュベクオ群 85例(13.0%)、プラセボ群 61例(9.4%) *2:ニュベクオ群 91例(14.0%)、プラセボ群 68例(10.5%)
注目すべき有害事象 [因果関係を問わない事象]
データカットオフ:2021年10月25日
【試験薬投与期間中央値】 ニュベクオ群:41.0ヵ月、プラセボ群:16.7ヵ月
| 有害事象*1、n(%) MedDRA Ver.25.0 | ニュベクオ群(ニュベクオ+ADT+ドセタキセル)(n=652) | プラセボ群(プラセボ+ADT+ドセタキセル)(n=650) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 全グレード | グレード3 | グレード4 | グレード5 | EAIR全グレード (100人年) | 全グレード | グレード3 | グレード4 | グレード5 | EAIR全グレード (100人年) | |
| 疲労/無力症 | 320(49.1) | 12(1.8) | 0 | 0 | 18.5 | 322(49.5) | 15(2.3) | 0 | 0 | 26.8 |
| 骨折(病的骨折を除く)*2 | 50(7.7) | 10(1.5) | 0 | 0 | 2.9 | 33(5.1) | 15(2.3) | 0 | 0 | 2.7 |
| 転倒(事故含む) | 45(6.9) | 2(0.3) | 0 | 0 | 2.6 | 33(5.1) | 2(0.3) | 0 | 0 | 2.7 |
| 血管拡張・潮紅 | 137(21.0) | 0 | 0 | 0 | 7.9 | 141(21.7) | 1(0.2) | 0 | 0 | 11.7 |
| 乳房障害/女性化乳房*3 | 21(3.2) | 0 | 0 | 0 | 1.2 | 10(1.5) | 0 | 0 | 0 | 0.8 |
| 皮膚障害*4 | 113(17.3) | 8(1.2) | 1(0.2) | 0 | 6.5 | 89(13.7) | 1(0.2) | 0 | 0 | 7.4 |
| 高血圧*3, 5 | 90(13.8) | 42(6.4) | 1(0.2) | 1(0.2) | 5.2 | 63(9.7) | 22(3.4) | 1(0.2) | 0 | 5.2 |
| 心臓障害 | 73(11.2) | 10(1.5) | 3(0.5) | 4(0.6) | 4.2 | 77(11.8) | 15(2.3) | 1(0.2) | 3(0.5) | 6.4 |
| 不整脈*3 | 53(8.1) | 2(0.3) | 2(0.3) | 1(0.2) | 3.1 | 56(8.6) | 4(0.6) | 0 | 2(0.3) | 4.7 |
| 冠動脈障害*3 | 21(3.2) | 7(1.1) | 1(0.2) | 2(0.3) | 1.2 | 13(2.0) | 7(1.1) | 0 | 0 | 1.1 |
| 心不全*3 | 4(0.6) | 1(0.2) | 0 | 1(0.2) | 0.2 | 13(2.0) | 5(0.8) | 1(0.2) | 1(0.2) | 1.1 |
| 糖尿病・高血糖 | 100(15.3) | 23(3.5) | 1(0.2) | 0 | 5.8 | 94(14.5) | 29(4.5) | 6(0.9) | 0 | 7.8 |
| 精神的機能障害*3 | 26(4.0) | 0 | 0 | 0 | 1.5 | 15(2.3) | 0 | 0 | 0 | 1.2 |
| 抑うつ性気分障害*3 | 23(3.5) | 0 | 0 | 0 | 1.3 | 26(4.0) | 0 | 0 | 0 | 2.2 |
| 脳虚血 | 9(1.4) | 3(0.5) | 0 | 0 | 0.5 | 8(1.2) | 2(0.3) | 0 | 1(0.2) | 0.7 |
| 脳出血・頭蓋内出血 | 6(0.9) | 2(0.3) | 2(0.3) | 2(0.3) | 0.3 | 1(0.2) | 1(0.2) | 0 | 0 | 0.1 |
| 痙攣発作 | 4(0.6) | 0 | 0 | 0 | 0.2 | 1(0.2) | 0 | 0 | 0 | 0.1 |
| 体重減少 | 24(3.7) | 0 | 0 | 0 | 1.4 | 36(5.5) | 1(0.2) | 0 | 0 | 3.0 |
EAIR:曝露期間で調整した発現割合 グレードはNCI-CTCAE Ver 4.03に準じる。
- *1: 有害事象のグループ化には、MedDRA 器官別大分類(SOC)、高位グループ語(HLGT)、高位語(HLT)及びMedDRA ラベリング分類(MLG)を用いた。
- *2: MedDRA用語のあらゆる骨折および脱臼、四肢骨折および脱臼、骨盤骨折および脱臼、頭蓋骨骨折、顔面骨骨折および脱臼、椎体骨折および脱臼並びに胸郭骨折および脱臼を組み合わせた用語。
- *3: MedDRAの高位グループ語。
- *4: MedDRA用語の発疹、斑状丘疹状皮疹、薬疹、そう痒性皮疹、紅斑性皮疹、斑状皮疹、丘疹性皮疹、毛孔性皮疹、膿疱性皮疹及び小水疱性皮疹を組み合わせた用語。
- *5: 事後解析のデータを含む。
試験薬(ニュベクオ/プラセボ)の投与状況
- ニュベクオ群の29.1%、プラセボ群の24.5%で試験薬の用量調節が行われました。
- 予定投与量に対する実投与量の割合(RDI)の平均値は、ニュベクオ群で97.2%、プラセボ群で98.5%でした。
| ニュベクオ群 (ニュベクオ+ADT+ドセタキセル) (n=652) |
プラセボ群 (プラセボ+ADT+ドセタキセル) (n=650) |
||
|---|---|---|---|
| 投与期間 中央値 | 41.0ヵ月 | 16.7ヵ月 | |
| 用量調節 なし,n | 462/652(70.9%) | 491/650(75.5%) | |
| 用量調節 あり*1,n 減量例,n 休薬例,n 再増量例*2,n |
190/652(29.1%) 73 169 29 |
159/650(24.5%) 42 139 24 |
|
| 予定投与量に対する 実投与量の割合 (RDI)*3 |
平均値 | 97.2% | 98.5% |
| 中央値 | 100.0% | 100.0% | |
- *1: 治療開始から治療終了までの変更のみが含まれる。用量調節には、休薬/投与延期及び減量が含まれる。
また、患者の過誤により投与を1回スキップした場合も用量調節に含まれる。 - *2: 試験薬の減量を行った患者のうち、再増量をした患者数
- *3: 予定投与量に対する実投与量の割合は、投与の中断や減量の期間を含めて算出した
ドセタキセルの投与状況
- ドセタキセルの総投与サイクル中央値は両群ともに6サイクルであり、ニュベクオ群の87.6%、プラセボ群の85.5%が全6サイクルのドセタキセル投与を受けました。
- ニュベクオ群の60.0%、プラセボ群の62.9%でドセタキセルの用量調節が行われました。
| ニュベクオ群 (ニュベクオ+ADT+ドセタキセル) (n=652*1) |
プラセボ群 (プラセボ+ADT+ドセタキセル) (n=650*1) |
||
|---|---|---|---|
| 総投与サイクル 中央値*2 | 6サイクル | 6サイクル | |
| 6サイクル投与例,n | 571/652(87.6%) | 556/650(85.5%) | |
| 用量調節 なし,n | 261/652(40.0%) | 241/650(37.1%) | |
| 用量調節 あり,n 減量例,n 休薬例,n |
391/652(60.0%) 135 348 |
409/650(62.9%) 129 356 |
|
| 実際に投与したサイクルにおける 予定投与量に対する 実投与量の割合*3 |
平均値 | 96.0% | 95.8% |
| 中央値 | 98.6% | 98.5% | |
- *1: ニュベクオ群で10例、プラセボ群で13例の患者がドセタキセル投与を受けなかった。
- *2: ニュベクオ群 n=642 プラセボ群 n=637
- *3: ニュベクオ群 n=638 プラセボ群 n=635
ニュベクオ+ADT+ドセタキセルは、mCSPC患者のOS延長に貢献できる治療選択肢です
1. mCSPC患者1,305例を対象としたARASENS試験におけるOS、CRPCとなるまでの期間等の結果は以下のとおりでした
ニュベクオ群でOSの有意な延長が認められ、ニュベクオにより死亡リスクが32.5%低下しました*1。
48ヵ月時点での生存率は、ニュベクオ群で62.7%、プラセボ群で50.4%でした(主要評価項目、検証的解析結果) 。
ニュベクオ群でCRPCとなるまでの期間の有意な延長が認められ*2 、副次評価項目の7項目のうち5項目において、ニュベクオの有効性が示されました(副次評価項目)。
PSA奏効率(その他の評価項目、無作為割付け後12ヵ月時点)
- • 相対的PSA奏効率(90%以上低下):ニュベクオ群84.3%、プラセボ群57.5%
- • 絶対的PSA奏効率(PSA値 0.2ng/mL未満):ニュベクオ群60.2%、プラセボ群26.1%
2. ARASENS試験における安全性の結果は以下のとおりでした
全グレードの有害事象(因果関係を問わない事象)発現率*3:ニュベクオ群99.5%、プラセボ群98.9%
グレード3/4の有害事象発現率:ニュベクオ群66.4%、プラセボ群63.5%
試験薬の投与中止に至った有害事象:ニュベクオ群13.7%、プラセボ群10.6%
ニュベクオ群でプラセボ群に比べて発現率が3%以上高い有害事象:貧血、食欲減退、高血圧、四肢痛、AST増加
3. ARASENS試験における試験薬およびドセタキセルの投与状況は以下のとおりでした
試験薬のRDI平均値:ニュベクオ群97.2%、プラセボ群98.5%
ドセタキセルの総投与サイクル中央値は両群ともに6サイクルであり、ニュベクオ群の87.6%、プラセボ群の85.5%が全6サイクルのドセタキセル投与を受けました。
- *1 ハザード比 0.675〔95%CI: 0.568~0.801〕、p<0.0001(層別log-rank test、有意水準:片側0.025)
- *2 ハザード比 0.357〔95%CI: 0.302~0.421〕、p<0.0001(層別log-rank test、有意水準:片側0.025)
- *3 本安全性データを当局へ提出後、当局からの照会事項に基づき、プラセボ群の2例[悪心、背部痛 各1例](いずれも因果関係は否定された)を追加報告している。
層別化因子:病変の進展度(M1a vs M1b vs M1c)、ALP(ULN 以上 vs ULN 未満)
